بهنظر میرسد اکثر سرطانها بهعلت تغییراتی که در طول عمر فرد مبتلا رخ میدهد، بوجود میآیند. برخی از سرطانها وابسته به ژنهای معیوب (جهشیافته) هستند که از والدین فرد به ارث می رسند. در واقع، این سرطان نیست که به ارث میرسد، بلکه ژنهای معیوب هستند که باعث میشوند احتمال ابتلا به سرطانهای خاص بیشتر شود. خانوادههایی که دارای این ژنها هستند احتمال ابتلا به سرطان در آنها بسیار بیشتراست.
فهرست جهشهای شایع ژنتیکی در سرطانهای فامیلی و توارثی از مؤسسه ملی سرطان، شامل جهشهایی است که بر چندین ژن سرکوبکننده تومورهای مختلف تأثیر میگذارند. سرطانهای ناشی از تغییرات ارثی در ژنهای سرکوبکننده تومور شامل سرطان پستان، سرطان تخمدان، سرطان پروستات، لوسمی، سرطان پانکراس و سرطان روده بزرگ است (۱) (۲) (۳).
نکته: فقط قسمتی از هریک از این سرطانها با جهشهای ارثی مرتبط است بقیه موارد اسپورادیک بوده و ارثی نیستند.
درحال حاضر بسیاری از سرطانها برای بررسی درمان با داروهای موردنظر بهطور مرتب تحت آزمایش قرار میگیرند، این دسته شامل برخی از موارد مرتبط با سندرمهای سرطان ارثی و فهرست رو به رشدی از سرطانهای دیگر است.
از آنجا که ابتلا به سرطانهای نامبرده در لیست زیر، میتواند بهعلت جهشهای ژنی ارثی باشد، بسیاری از افراد مبتلا به آنها برای بررسی وجود ژن موردنظر، انتخاب میشوند (۴) (۵).
- سرطان ارثی پستان
- سندرم لی– فرامینی
- سندرم کاودن
- سندرم لینچ
- پولیپوز آدنوماتوز فامیلی
- رتینوبلاستوم
- نئوپلازی اندوکرین چندگانه
- سندرم فون هیپل– لیندو
در زیر شرح مختصری از موارد بهتر شناختهشده سرطانهای ارثی داده خواهد شد:
سرطان پستان و تخمدان ارثی
ژنهای: BRCA۱ ,BRCA۲
انواع سرطانهای مرتبط: سرطانهای پستان و تخمدان در خانمها و سرطانهای دیگری از قبیل سرطانهای پروستات، پانکراس و سرطان پستان در مردان
BRCA۱ و BRCA۲ ژنهای سرکوبگر تومور هستند که محصولات پروتئینی آنها مسئول جلوگیری از تقسیم سلولهای کنترلنشده میباشند. بهطور خاص، پروتئینهای BRCA۱ و BRCA۲ در بهبود آسیب به DNA و کنترل ژنهای دیگر دخیل هستند. علت اینکه جهش در این ژنها بهطور خاص به سرطان پستان و تخمدان مرتبط است، کاملاً مشخص نیست، اما ممکن است به هورمون استروژن مربوط باشد. سلولهای پستان و تخمدان برای تکثیر به هورمون استروژن وابستهاند و به تغییر مقادیر استروژن واکنش نشان میدهند (برای مثال، مقادیر استروژن تحت تاثیر چرخه قاعدگی و بلوغ است). تقسیم سریع ناشی از استروژن ممکن است منجر به افزایش جهش در این ژنها و رشد بعدی سرطان گردد (۶) (۷) (۸).
سندرم لی– فرامینی
ژن: TP۵۳
انواع سرطانهای مرتبط: سرطان پستان، سارکوم بافت نرم، استئوسارکوم (سرطان استخوان)، لوسمی، تومورهای مغزی، کارسینوم آدرنوکورتیکال (سرطان غدد فوق کلیه) و سرطانهای دیگر
ژن TP۵۳ (یا ژن P۵۳) یک پروتئین بسیار مهم سرکوبگر تومور را کپی میکند. این ژن در بسیاری از فعالیتهای جلوگیریکننده از تقسیم غیرقابل کنترل سلولی دخالت دارد. فعالیتهای مهم تنظیمشده توسط P۵۳ عبارتند از: ترمیم DNA، مرگ سلولی (آپوپتوز) و کنترل چرخه سلولی. به این ژن “دروازهبان” میگویند بدین لحاظ که کار آن در نهایت محافظت بدن از تشکیل تومور است. جهشهای ارثی در TP۵۳ باعث مستعد شدن فرد به انواع سرطان میشود، زیرا این ژن یکی از اصلیترین عوامل دفاعی بدن در برابر فعالیتهای سرطانزا است (۹) (۱۰) (۱۱) (۱۲).
سندرم کاودن (سندرم تومور هامارتوم P TEN)
ژن:TENP (همولوگ فسفاتاز و تنسین)
انواع سرطانهای مرتبط: پستان، تیروئید، آندومتر (پوشش رحم) و سرطانهای دیگر
PTEN نیز یک ژن سرکوبگر توموراست. درست مانند TP۵۳ اگر PTEN هم معیوب باشد، سلولها ممکن است به تقسیم شدن ادامه دهند، حتی ممکن است تغییرات ایجادکننده سرطان رخ بدهند. بهطور خاص، محصول PTEN، پاسخ مثبت یا منفی سلولها به پیامهای تقسیم شدن و یا تحت آپوپتوز (نسخه خودکشی سلولی) قرار گرفتن را کنترل میکند. اگر تنظیم این پیامها غیرفعال شود، سلولها ممکن است بیوقفه و خارج از کنترل تقسیم شوند و تومور تشکیل شود. جهشهای PTEN مسئول سندرم کاودن که شامل شکل گرفتن بسیاری از هامارتومها که رشد غیربدخیمی هستند، میباشند. جهشها همچنین منجر به افزایش ریسک سرطان پستان، سرطان تیروئید و سرطان آندومتر (سرطان پوشش رحم) میشوند (۱۳) (۱۴) (۱۵).
سندرم لینچ (سرطان کولورکتال غیرپولیپوز ارثی)
ژنها:MSH۲ ،MLH۱ ، MSH۶،PMS۲ ،EPCAM
انواع سرطانهای مرتبط: سرطانهای کولورکتال، آندومتر، تخمدان، لگن کلیوی، پانکراس، روده کوچک، کبد و مجرای صفراوی، معده، مغز و پستان
ژنهای درگیر در سندرم لینچ، ژنهای بازسازی نامتقارن DNA هستند. پروتئینهایی که توسط این ژنها کدگذاری شدهاند مسئول اصلاح اشتباهاتی هستند که هنگام کپی شدن (تکثیر) DNA بوجود آمدهاند. هنگامی که این ژنها معیوب باشند، پروتئینها بهدرستی قادر به بازسازی DNA نخواهند بود. اغلب سرطانهای مرتبط با سندرم لینچ رامیتوان با “بیثباتی ریزماهوارهها” شناسایی کرد. ریزماهواره اصطلاح توالی تکرارشونده DNA مانندCGCGCGCGG یا TATATATAT است. ژنوم انسانی از این نوع توالی تکرارشونده زیاد دارد. بیثباتی ریزماهوارهها یعنی جهشهایی که بهطور خاص در این توالیهای تکرارشوندهی DNA رخ میدهند. معمولا ًبرخی از تکرارها از بین رفته و یا اضافه میشوند (مثلاً CAGCAGCAG به CAGCAG تبدیل میشود).
تغییرات در توالیهای تکرارشونده میتوانند بر روی پایداری DNA تأثیر گذاشته و منجر به ایجاد انواع سرطانها شوند (۶) (۷) (۸).
پولیپوز آدنوماتوز فامیلی
ژن: APC (کولیت پولیپوز آدنوماتوز)
انواع سرطانهای مرتبط: سرطان کولورکتال، تومورهای روده کوچک، مغز، معده، استخوان، پوست و بافتهای دیگر. همچنین با رشدهای (پولپهای) غیرسرطانی (خوشخیم) روده بزرگ و روده کوچک همراه است.
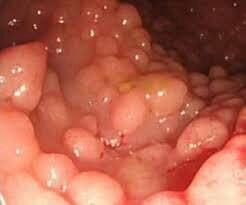
APC یک ژن سرکوبگر توموراست که چگونگی تقسیم سلولی، چگونگی چسبیدن سلولها به یکدیگر و چگونگی حرکت سلولی را کنترل میکند. این ژن همچنین در تشخیص آسیب DNA سهیم است و با سایر پروتئینهای دخیل در ارتباط بین سلولها همراهی میکند. بسیاری از جهشهای مختلف در APC بهعنوان علت ایجاد پولیپوز آدنوماتوز فامیلی شناخته شدهاند، یک بیماری که با پیدایش صدها پولیپ توأم است. بسیار محتمل است که حداقل یکی از پولیپها در برخی مواقع در طول زندگی بیمار بتواند سرطانی شود. پروتئین معیوب APC نیز میتواند منجر به ایجاد تومورهای دسموئید که تومورهای خوشخیم ضخیم بافت همبند هستند، بشود (۱۶) (۱۷) (۱۸) (۱۹).
رتینوبلاستوم
ژن: RB۱ (رتینوبلاستوم)
انواع سرطانهای مرتبط: سرطان چشم (سرطان شبکیه)، پینهآلوم (سرطان غده صنوبری)، استئوسارکوم، ملانوم و سارکوم بافت نرم
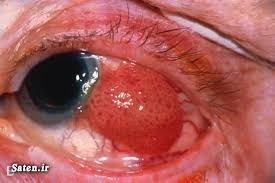
ژن RB۱ که یک ژن سرکوبگر است، پروتئین Rb را کد میکند. پروتئین Rb مسئول متوقف کردن تقسیم سلولی در شرایط نامطلوب است (بهطور مثال در هنگام آسیب DNA که بایستی بازسازی شود، یا زمانی که سلول بهنوعی تحت فشار است). این ژن در کنترل سایر پروتئینهایی که در تکثیر DNA، آپوپتوز و بلوغ سلولی (تمایز) دخیل هستند، نقش دارد. هنگامی که جهشی در ژن RB۱ رخ دهد، پروتئین Rb ممکن است فعالیت نکند، در این صورت رشد سلولی بیقاعده خواهد شد. به دلایلی که بهطور کامل مشخص نیست، تغییرات در RB۱ باعث ایجاد سرطان در چشم بهویژه در شبکیه میشود. زمانی که یک کپی جهشیافته از ژن رتینوبلاستوم به ارث برده شود (باعث ایجاد سرطانهایی به نام رتینوبلاستوم ژرمینال یا فامیلیال شود)، ژن جهشیافته در هر سلول بدن یافت میشود و فرد را بهشدت به انواع دیگر سرطان، در درجه اول سرطان غده صنوبری، استخوانها، بافت نرم و پوست، حساس میکند (۲۰) (۲۱) (۲۲) (۲۳) (۲۴).
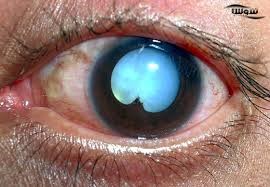
مولتیپل اندوکرین نئوپلازی تیپ ۱ (سندرم ورمر)
ژن: MEN۱
انواع سرطانهای مرتبط: تومورهای درونریز پانکراس و (معمولاً خوشخیم) تومورهای پاراتیروئید و غده هیپوفیز
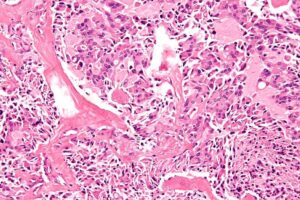
MEN۱ یک پروتئین سرکوبگر تومور به نام منین را کپی میکند. عملکرد دقیق منین ناشناخته است، اما بهنظر میرسد که در تنظیم تقسیم سلولی، بازسازی DNA و آپوپتوز دخیل باشد. بیش از هزار جهش مختلف درژن MEN۱ موجب ایجاد مولتیپل اندوکرین نئوپلازی تیپ ۱ میشوند. تیپ ۱ ژن MEN در رشد تومور داخل غدد درونریز (غدد تولیدکننده هورمون) مداخله میکند. غدد اندوکرینی که غالباً تحت تأثیر مولتیپل نئوپلازی تیپ ۱ هستند عبارتند از غده پاراتیروئید، غده هیپوفیز و پانکراس. معمولاً جهش در MEN۱ منجر به تولید نسخه کوتاهشده پروتئین منین میشود که ناپایداراست و بهراحتی تجزیه میشود. وقتی چنین چیزی اتفاق میافتد، یک کپی از ژن MEN۱، پروتئین عملکردی منین را تولید نمیکند. اگر جهش در نسخه دوم (که در غدد درونریز عادی است، اگرچه دلیل آن ناشناخته است)، رخ دهد، سلول نمیتواند بههیچوجه منین مناسبی را تولید کند و منجر به تقسیم غیرقابل کنترل سلول و سرطان میگردد (۲۵) (۲۶) (۲۷) (۲۸) (۲۹) (۳۰).
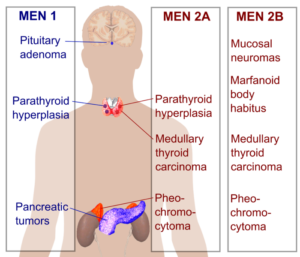
مولتیپل اندوکرین نئوپلازی تیپ ۲
ژن:RET
انواع سرطانهای مرتبط: سرطان تیروئید مدولار و فئوکروموسیتوم (تومور خوشخیم غده فوق کلیه)
ژن RET یک پروتوانکوژنی است که در پیامرسانی سلولی دخالت دارد و غشای سلولی را دربر میگیرد و بهعنوان گیرندهای برای پیامهایی که به پاسخ سلولها به تغییرات محیطی اطرافشان کمک میکنند، عمل مینماید. MEN۲ به سه زیرگونه تقسیم میشود: MEN۲A ,MEN۲B و کارسینوم تیروئید مدولاری فامیلی (FMTC). بیشتر جهشها در ژن RET که باعث ایجاد MEN۲ میشوند جهشهای بسیار کوچک (نقطهای) هستند که باعث تغییر در فقط یک اسیدآمینه پروتئین میشوند. بسیاری از این جهشها با سرطان تیروئید مدولاری ارثی (فامیلی) مرتبط هستند (۲۷) (۲۸) (۳۰) (۳۱) (۳۲).
سندرم فون هیپل– لیندو
ژن: VHL
انواع سرطانهای مرتبط: سرطان کلیه و تومورهای متعدد غیر سرطانی از جمله فئوکروموسایتوم
ژن VHL به همراه سایر پروتئینها کمپلکس VCB- CUL۲ را تشکیل میدهند. این کمپلکس باعث میشود که سایر پروتئینهای درون سلول، زمانی که آسیب دیدهاند و یا دیگر نیازی به آنها نباشد، شکسته شوند. یکی ازتارگتهای این کمپلکس، فاکتور القاءشده با هیپوکسی ۲-alpha(HIF۲a) است. HIF۲a پاسخ بدن به تغییرات سطح اکسیژن را با کنترل تقسیم سلولی و تشکیل رگهای خونی جدید و گلبولهای قرمز انطباق میدهد.
وقتی که سطح اکسیژن نرمال باشد، کمپلکس VCB- CUL۲، فاکتور HIF۲a را متوقف میکند. زمانی که VHL جهش پیدا کند، کمپلکس VCB- CUL۲ بهدرستی عمل نخواهد کرد و نمیتواند HIF۲a یا پروتئینهای دیگری که آسیب دیدهاند و یا موردنیاز نیستند را تخریب کند، در این صورت ممکن است HIF۲a باعث تحریک تقسیم بیش ازحد سلولی و تشکیل عروق خونی شود که میتواند منجر به ایجاد تومور و کیست که هر دو مشخصه سندرم فون هیپل- لیندو هستند، گردد
فراپژوهش