شناسایی سریع و دقیق عامل میکروبی جهت درمان افراد مصدوم و کنترل انتقال آلودگی به افراد سالم مهم میباشد. امروزه در آزمایشگاههای میکروب شناسی پیشرفته در دنیا جهت تشخیص عوامل میکروبی از روشهای رایج و سنتی مانند آزمایش مستقیم، کشت و روشهای ایمنولوژی کمتر استفاده شده و از روشهای مولکولی مانند (PCR، وسترن بلات، ایمنوبلات، ایمنو اسی، ایمنو فلورسانس و غیره) استفاده میکنند. روشهای تشخیص مولکولی، جداسازی و شناسائی پاتوژنها را بر اساس شاخصهای مولکولی خاص انجام میدهند. این سیستمها دارای سرعت، حساسیت و اختصاصیت بالا نسبت به روشهای معمول در آزمایشگاه میباشند. همچنین بدلیل اینکه بیشتر سنجشهای مولکولی امروزه، به صورت اتوماتیک قابل دسترس میباشند، لذا نسبت به روشهای سنتی در آزمایشگاه سادهتر انجام میشوند. اصولاً از روشهای مولکولی در آزمایشگاه میکروب شناسی برای شناسائی عوامل غیر قابل کشت (مانند ویروس هپاتیت B)، عوامل با رشد آرام و سختگیر (مانند مایکوباکتریم توبرکلوزیس)، عوامل عفونی که قدرت بیماریزائی بالائی داشته و در کشت دادن امکان آلودگی کارکنان آزمایشگاه وجود دارد (مانند فرانسیسلا تولارنسیس و گونههای بروسلا)، عواملی که در نمونه به مقدار کم وجود دارند (مانند HIV در افراد سرم منفی) و نمونه مورد آزمایش بسیار کم باشد (مایعات مغز- نخاعی و نمونههای جرم شناسی)، متمایز کردن عوامل مشابه از نظر آنتیژنیک، ارگانیسمهای غیرزنده (ارگانیسمهایی که سبب تشکیل کمپلکس ایمن در بدن میشوند) استفاده میشود.
بطورکلی روشهای شناسایی سریع عوامل میکروبی عبارتند از:
۱- جداسازی و شناسایی عامل میکروبی از طریق کشت دادن (معمولاً یک یا دو روز برای بعضی از عوامل طول میکشد).
۲- شناسایی سموم توسط دستگاههای اسپکتروسکوپی جرمی، تلقیح به حیوانات آزمایشگاهی و یا روشهای دیگر.
۳- تشخیص ایمنولوژیک، شناسایی آنتیبادیها (ایمونوگلوبینهای خاص از نوع IgM که ممکن است در عرض سه روز پس از عفونت حاصل گردد).
۴- شناسایی ضایعات ژنتیکی از طریق شاخصهای تشخیصی DNA probe
۵- شناسایی محصولات متابولیکی عامل عفونتزا و مواد سمی در نمونههای بالینی
تشخیص مولکولی عوامل میکروبی میتواند بر اساس Central dogma بیولوژی مولکولی انجام شود. بر اساس این Central dogma، DNA همانندسازی میکند و RNA از روی DNA نسخه برداری شده و در نهایت RNA نسخه برداری شده (mRNA) در سطح ریبوزوم قرار میگیرد و به پروتئین ترجمه میگردد ( شکل ۱). لذا با توجه به این Central dogma اهداف مولکولی در تشخیص میکروبها عبارتند از:
۱- DNA: شامل ژنوم کامل- ژن تکی و سکانس کوتاه
۲- RNA: به خصوص در RNA ویروسها. این روش خیلی بندرت در باکتریها بکار میرود چرا که RNA در باکتریها پایدار نمیباشد، هر چند در ریبوتایپینگ از RNA استفاده میگردد.
۳- پروتئین: شامل پروتئینهای ایمونولوژیک میباشد و اصولاً با روشهای سرولوژی و (Sodium dodecyl sulfate Polyacrylamide gel electrophoresis)SDS-PAGE قابل شناسائی میباشند.
۴- دیگر موارد: مانند LPS باکتریها که با روشهای سرولوژی و SDS-PAGE قابل شناسائی میباشد.

شکل ۱: dogma مرکزی بیولوژی مولکولی
بطور کلی تکنیکهای مولکولی یا DNA یا RNA را شناسائی کرده و کمتر پروتئینها و مواد دیگر را شناسائی میکنند. امروزه این تکنیکها در آزمایشگاههای تشخیصی و تحقیقاتی بسیار استفاده شده و روزبروز در حال توسعه و تکامل میباشند. تشخیصهای مولکولی بر اساس روش تشخیص به چهار دسته کلی تقسیم میشود:
۱- تکنیکهای تشخیصی که نیاز به آمپلی کردن ژن مورد نظر ندارند مستقیماً بدون آمپلی کردن سکانس خاص، میکروب را شناسائی میکنند. مانند In situ hybridization,FISHو غیره.
۲-تکنیکهای تشخیصی که ژن مورد نظر را آمپلی میکنند مانند انواع مختلف PCR، TBA و غیره.
۳ – تکنیکهای تشخیصی که پروپ نشاندار را آمپلی میکنند مانندLCR ، PCR/OLA و غیره.
۴- تکنیکهای آنالیز و جداسازی بعد از آمپلی کردن مانند ژل الکترفوز، PFGE، کالریمتری، کیمولومینانس و غیره.
در ادامه بحث به جزئیات بعضی از این روشهای سریع مولکولی بکار رفته در آزمایشگاههای مدرن میکروب شناسی جهت شناسایی عوامل میکروبی اشاره خواهد شد.
۱- Fluorescence In Situ Hybridization (FISH)
در این روش با کمک شناساگر از نوع ۱۶S -rRNA یا ۲۳S -rRNA نشاندار و میکروسکوپ فلورسانس برای جداسازی مستقیم باکتری از نمونههای کلینیکی مانند خون یا بافت یا بعد از کشت روی محیطهای غنی استفاده میشود (شکل ۲). این روش برای جداسازی میکروبهای سخت رشد مانند یرسینا پستیس و بارتونلاها بسیار مفید میباشد و میتوان چندین گونه از باکتریها را به طور همزمان با استفاده از رنگهای فلورسانت مختلف شناسائی کرد. این روش از فیکس کردن نمونه روی لام، هیبرید کردن نمونه با پروپ و بررسی هیبریدها بوسیله میکروسکوپ فلورسانس حدود ۱-۲ ساعت طول میکشد.
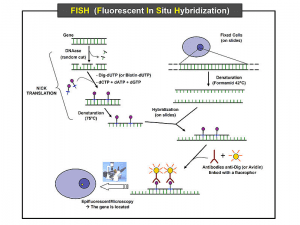
شکل ۲: مراحل مختلف تکنیک FISH
۲- شناسائی بر اساس سکانس کردن
اگر ارزیابی پاتوژن باکتری خاص مشکل میباشد یا هنگامی که چندین عامل به عنوان عامل بیماری مطرح باشند بایستی از روشی که قدرت شناسائی عوامل در محدوده وسیع را دارند استفاده شود. اهداف عمومی مانند ژنهای۱۶S-rRNA یا نواحی پراکنده ژن ۱۶S –۲۳ S rRNA برای شناسائی باکتریها بسیار مفید میباشد به خصوص که این باکتریها با استفاده از روشهای عمومی مشکل جدا میشوند. سکانس کردن آن ۱۶S -rRNA اغلب برای جداسازی پاتوژنها از نمونههای کشت منفی مورد استفاده قرار میگیرد. امروزه سکانسهای ژن ۱۶S -rRNA برای تایپینگ کردن بعضی از پاتوژنها مورد استفاده قرار میگیرد. این روش بسیار ارزان و سریعتر از روشهای بیوشیمیائی میباشد. میتوان ژنهای ۱۶S -rRNA را با کمک PCR آمپلی کرد و بعد آن را سکانس کرد. لازم بذکر است از اهداف دیگری که میتوان برای سکانس کردن آن و شناسائی باکتریها مورد استفاده قرار داد پروتئینهای شوک گرمائی (HSP-۶۵ )یا پروتئینهای شوک سرمائی میباشد.
۳- الکتروفورز ژل آگارز
الکتروفورز در ژل آگارز در حرارت اتاق در حالت افقی، روشی استاندارد برای جداسازی قطعات DNA است. ژل آگارز در مقایسه با ژل پلی آکریل آمید قدرت تفکیک کمتری داشته ولی محدوده جداسازی بیشتری دارد. قطعات DNA به اندازه ۵۲۰۰جفت باز تا ۶۰ کیلو باز را میتوان در غلظتهای متفاوت ژل آگارز از هم جدا نمود. در جدول( ۱) مقدار آگارز موردنیاز در ژل برای جداسازی ملکولهای خطی DNA نشان داده شده است. برای ساختن ژل آگارز ابتدا پودر آگارز را در بافر مناسبی که دارای EDTA است ریخته و حرارت میدهند تا محلول شفاف و روشنی حاصل گردد. این عمل را میتوان با گذاشتن مخلوط بافر و آگارز در ماکروویو انجام داد. سپس، محلول را تا ۶۵ درجه سلسیوس سرد کرده و بعد، به درون قالب ژل الکتروفورز یا روی لام میریزند تا سفت گردد. قبل از ریختن آگارز یک شانه پلاستیکی را روی لام یا درون قالب ژل آگارز قرار میدهند. این عمل بدان جهت انجام میشود تا دندانههای شانه، چاهکهای (حفرههای) کوچکی را در آگارز قبل از سفت شدن تشکیل دهند. برای تعیین فاصله دندانههای شانه تا کف ظرف قالب میتوان لامی را در زیر دندانههای شانه قرار داده و پس از تنظیم، لام را برداشته و آگارز را اضافه کرد. زمانی که آگارز سفت شد، ماتریکس ضخیمی را تشکیل میدهد که به مقدار غلظت آگارز بستگی دارد. سپس DNA مورد نظر به درون چاهکها وارد میشود. هنگامی که جریان برق از میان ژل عبور کند DNA که بار منفی در pH خنثی دارد به طرف کاتد حرکت میکند. میزان مهاجرت DNA در ژل به عوامل زیر بستگی دارد:
– اندازه DNA: مولکولهای بزرگتر، آهستهتر از ملکولهای کوچکتر در زمان یکسان حرکت میکنند.
– غلظت آگارز: اندازههای مساوی از ملکولهای خطی DNA با سرعتهای متفاوتی در غلظتهای مختلفی از ژل آگارز حرکت میکنند (جدول۱).
– شکل فضائی DNA: ملکولهای سوپرکویل (ابرمارپیچی) حلقوی و خطی DNA با اندازههای یکسان در ژل با غلظت مشخص با سرعتهای متفاوتی حرکت میکنند.
اشکال مختلف به ترتیب متفاوتی حرکت میکنند که به شرایط موجود بستگی دارد. هنگامی که ژل سفت شد و نمونهها به آن اضافه گردید، دستگاه تنظیم برق را وصل کرده تا جریان برق برقرار گردد. ضروری است که جهت صحیح الکترودها رعایت شود وگرنه، DNA در جهت اشتباه حرکت خواهد کرد. توجه شود که نمونهها در آن قسمتی که به قطب منفی و بخش مخالف آن به قطب مثبت وصل شده است قرار گیرد.
جدول (۱): مقادیر آگارز موردنیاز در ژل برای جداسازی ملکولهای خطی DNA
| مقدار آگارز در ژل (W/V%) | جداسازی ملکولهای خطی (kbp)DNA |
| ۰/۳ | ۵ تا ۶۰ |
| ۰/۶ | ۱ تا ۲۰ |
| ۰/۷ | ۰/۸ تا ۱۰ |
| ۰/۹ | ۰/۵ تا ۷ |
| ۱/۲ | ۰/۴ تا ۶ |
| ۱/۵ | ۰/۲ تا ۳ |
| ۲ | ۰/۱ تا ۲ |
نمونه با مقداری از رنگ نشانگر مخلوط میشود. این رنگ از دو جزء تشکیل شده که در دادن اطلاعات تخمینی مبنی بر حرکت نمونه حاوی DNA در ژل مفید میباشد. یک جزء یعنی سیانول گزیلن (به رنگ سبز) در اندازهای حدود ۴kb حرکت میکند، در صورتی که جزء دیگر، بروموفنل بلو (رنگ آبی تیره) در اندازهای حدود ۴۰۰bp حرکت میکند. از این رنگها میتوان به عنوان راهنما برای زمان مناسب انجام آزمایش بر روی ژل استفاده نمود.
پس از انجام عمل الکتروفورز برای مشاهده DNA باید رنگآمیزی ژل با اتیدیوم بروماید (EtBR)یا ترکیبات دیگر انجام گیرد. این رنگ بین بازهای دو زنجیره DNA نفوذ میکند و در زیر نور اولتراویوله به رنگ قرمز- نارنجی فلورسنس مشاهده میگردد. مقدار اتیدیوم بروماید مورد استفاده به طول قطعه DNA بستگی دارد. بعد از انجام ژل الکتروفورز در محلول اتیدیوم بروماید رنگآمیزی شده و سپس با آب مقطر شستشو داده میشود تا رنگ متصل نشده به DNA که به درون ژل وارد شده حذف گردد. پس از آن از ژل توسط دستگاهGel Doucomentation
عکسبرداری میشود (شکل ۳).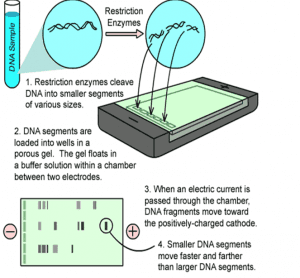
شکل۳: مراحل مختلف الکتروفورز
۴- Pulsed Field Gel Electrophoresis (PFGE)
تمام ملکولهای زنجیر مضاعف DNA خطی تا اندازه معینی به یک میزان از میان ژل آگارز عبور میکنند. بالاتر از این حد معین سرعت عبور ملکولهای DNA از میان ژل آگارز به اندازه DNA بستگی نداشته و به طور عمده به شدت جریان الکتریکی بستگی دارد. در عمل این ارتباط بدان معنی است که DNAهای بزرگتر از ۴۰kb نمیتوانند با استفاده از جریان ثابت الکتریکی در ژل آگارز افقی به سهولت از هم جدا شوند. این مشکل را میتوان با PFGE حل نمود که در آن، جریان الکتریکی به طور متناوب در دو جهت مختلف با زمانهای pulse از ۱/۰ تا ۱۰۰۰ ثانیه یا بیشتر تغییر میکند (شکل ۴). با این روش میتوان ملکولهای DNA موجود در نمونههای مشکوک تا حدود ۵ میلیون باز درازا را از هم جدا کرد. در این حالت زمانی که ملکول DNA نیاز دارد تا مسیر خود را در پاسخ به نوسان جریان الکتریکی تغییر دهد به اندازه ملکول DNA بستگی دارد، یعنی ملکولهای کوچکتر با سرعت بیشتری میتوانند تغییر جهت داده و بنابراین سریعتر از ملکولهای درازتر از میان ژل عبور میکنند (شکل ۵).

شکل ۴: جهات حرکت قطعات اسید نوکلئیک در PFGE
نکات زیر در انجام PFGE اهمیت بسیار داشته و باید به آنها توجه شود:
۱- فضای جداسازی: در اکثر سیستمهای PFGE، DNA در فضای کوچکی الکتروفورز میشود و وضوح آن به پیچیدگی نمونه بستگی دارد.
۲- قدرت میدان الکتریکی: قدرت میدان الکتریکی تأثیر مهمی بر جداسازی در روش PFGF دارد، به طوری که بین زمان جداسازی و مشاهده واضح DNA به اندازههای خاص به میدان الکتریکی خاصی نیاز میباشد.
۳- زمان Pulse: اولین تأثیر زمان pulse، تغییر میزان جداسازی است. از زمانهای pulse طولانیتر برای جداسازی DNAهای بزرگتر استفاده میشود.
۴- زاویههای تغییر جهت: جداسازی DNA در زاویههای ۱۶۵-۹۵ درجه برابر و مشابه است. با این حال، هر قدر زاویه کوچکتر باشد، حرکت DNA سریعتر میباشد. برای جداسازی DNAهای خیلی بزرگتر، زاویههای ۱۰۵-۹۶ درجه مورد نیاز است تا جداسازی DNA در کوتاهترین زمان ممکن به خوبی انجام شود.
۵- بافر: از بافرهای TBE و TBE استفاده میشود.
۶- آگارز: غلظت و نوع آگارز در جداسازی DNA مؤثر است. سریعترین حرکت و بهترین وضوح در ژلهایی بدست میآید که از Low electroendosomsis ساخته شده باشد. البته آگارز الکتروفورز معمولی برای PFGE مناسب است.
۷- دما: مناسبترین درجه حرارت برای انجام PFGE دمای ۱۴ درجه سلسیوس است.
کاربردهای PFGE و الکتروفورز ژل آگارز
اصولاً الکتروفورز معمولی و PFGE برای جداسازی و تعیین اندازه پلاسمیدهای بزرگ و DNAهای کروموزومی باکتریهای موجود در نمونههای مشکوک به عوامل میکروبی استفاده میشود. پس از الکتروفورز کردن نمونههای مشکوک و بدست آوردن باندهای مناسب، آنها با الگوهای مشخص مقایسه میشوند و عامل بیولوژیک تشخیص داده میشود.
از کاربردهای رایج دیگر الکتروفورز ژل آگارز و به خصوص PFGE در میکروب شناسی پزشکی، مقایسه اپیدمیولوژی باکتریهایی است که متعلق به یک خانواده هستند و در عفونتهای بیمارستانی و یا به دلیل تماسهای مستقیم انتقال مییابند. مقایسه باندهای مشاهده شده ممکن است دخالت سویههای جدا شده در اپیدمیها را نشان دهد.

شکل ۵: مراحل مختلف PFGE
۵- الکتروفورز ژل پلی اکریل آمید- سدیم دودسیل سولفات (SDS-PAGE)
برای آنالیز یک پروتئین خاص برای شناسایی یک عامل بیولوژیک (به خصوص سموم) در نمونههای مشکوک از الکتروفورز ژل پلی اکریل آمید استفاده میشود. در این روش، نمونه حاوی پروتئین مورد نظر در شرایطی قرار میگیرد که پروتئینها به زیر واحدهای پلیپپتیدی از هم تفکیک میشوند و غلظت پلیپپتیدهای متفاوت در مخلوط کاهش مییابد. بعد از مراحل تهیه، مخلوط پروتئینی بوسیله ژل پلی اکریل آمید، الکتروفورز میشود. سپس ژل را برای مشاهده پروتئینها، رنگآمیزی کرده و برای آنالیزهای بعدی مورد استفاده قرار میگیرد.
پروتئین مورد نظر بخشی از مخلوطی از پروتئینهای متفاوت است. برای اطمینان از جدا شدن پروتئینها به زیر واحدهای پلیپپتیدی و کاهش تجمع، معمولاً پروتئینهای موجود در مخلوط را تجزیه کرده، کاهش داده و یک دترجنت آنیونی (یعنی دودسیل سولفات سدیم ] SDS قلیائی[) به آن اضافه میشود. عمل تجزیه شدن نمونه را میتوان با جوشاندن به مدت کوتاه انجام داد. این عمل، ساختمانهای دومی، سومی و چهارتایی را که ممکن است وجود داشته باشد و همچنین معکوس شدن واکنشهای تصادفی پروتئین– پروتئین را که ممکن است رخ دهد تخریب میکند. عمل کاهش را میتوان با افزودن بتامرکاپتواتانول (BME) انجام داد. این معرف اتصالهای دی سولفیدی را با تخریب هر نوع ساختمان سومی که ممکن است موجود باشد حذف میکند و هم چنین مانع تشکیل هرگونه اتصالهایی میشود که ممکن است پلیپپتیدهای cross-link را از هم مجزا سازد.
افزودن SDS به پلیپیتید تخریب شده بار خالص منفی را به آن اعطا میکند. مقدار اتصال SDS به پروتئین، نسبت خطی با اندازه پروتئین دارد و به توالی اسید آمینه بستگی ندارد. چون SDS، بار خیلی بالایی دارد، پس به طور گستردهای اتصال مییابد و هر بار دیگر موجود در روی پروتئین نادیده گرفته میشود. بدین ترتیب، بار هر واحد وزن، پایدار بوده و حرکت در الکتروفورز به وزن مولکولی آن وابسته است.
این ژل با جوشاندن مخلوط پروتئین در بافری که حاوی BME،SDS میباشد حاصل میگردد. این امر منجر به ایجاد بار منفی در مخلوطی از کمپلکسهای پروتئین– SDS میشود. بافر همچنین دارای رنگ نشانگر میباشد که نشان دهنده مساحت حرکت انجام شده در روی ژل الکتروفورز میباشد. گلیسرول، ماده دیگری از بافر است که درload کردن نمونه به درون ژل کمک میکند. پروتئینهای موجود در مخلوط با عبور نمونه از میان شبکه آکریل آمید از هم جدا میگردند. این شبکه به عنوان غربالی، عبور پروتئینها را با حرکت آهسته فیزیکی آنها به تأخیر میاندازد. مولکولهای بزرگتر، آهستهتر از مولکولهای کوچکتر حرکت میکنند. مقدار درصد آکریل آمید در ژل همچنین بر روی میزان حرکت پروتئینها اثر دارد. محلول غلیظ پروتئینها، آهستهتر از غلظتهای پائین حرکت میکنند. در جدول(۲) میزان خطی جدایی پروتئینها با غلظتهای متفاوت آکریل آمید نشان داده شده است.
جدول (۲ ): نسبت غلظت آکریل آمید و جدا شدن پروتئینها
| غلظت آکریل آمید (%) | حدود جدا شدن خطی (کیلودالتون) |
| ۱۵ | ۱۲-۴۳ |
| ۱۰ | ۱۶-۶۸ |
| ۷/۵ | ۳۶-۹۴ |
| ۵ | ۵۷-۲۱۲ |
پلیپپتیدهای جدا شده توسطSDS-PAGE را میتوان با استفاده از رنگهای شیمیایی مشاهده کرد. پلیپپتیدها را میتوان به طور همزمان با مخلوطی از اسید استیک و متانول و رنگآمیزی کوماسی برلیانت بلو تثبیت نمود.
این رنگ برای برخی از اسیدهای آمینه (برای مثال، آرژینین و لیزین) و رنگآمیزی پلیپپتیدها در درون ژل اختصاصی است. رنگ اختصاصی را میتوان با رنگبری به مدت طولانی از ژل خارج کرد. با مقایسه باندهای حاصل با باندهای استاندارد شده در روی ژل (پروتئینهایی با اندازه شناخته شده) میتوان وزن مولکولی پلیپپتید مورد نظر را تخمین زد. عمل الکتروفورز بوسیله ژل با استفاده از مارکرهایی با وزن مولکولی مشخص که از قبل رنگآمیزی شده، تسهیل شده است، به طوری که مشاهده پایان عمل، قبل از اینکه پروتئین مورد نظر از ژل خارج شود، مقدور میباشد.
فراپژوهش