الکتروفورز ژل دناتورهکننده با شیب غلظت
تکنیک الکتروفورز ژل دناتورهکننده با شیب غلظت(DGGE)، کارآمدتر و حساستر از SSCP بوده و بیش از ۹۵% قدرت تشخیص جهشها را دارد، اما راهاندازی آن پیچیده و مشکل است. اساس این تکنیک بر این امر استوار است که در شرایط ثابت محیطی، نقطه ذوب یک قطعه از DNA دو رشتهای به توالی و ترکیب نوکلئوتیدهای آن بستگی دارد. دو قطعه DNA که در یک جفت باز با هم اختلاف دارند، ممکن است شرایط ذوب بسیار متفاوتی داشته باشند.
محصول PCR بر روی ژل پلیاکریلامید که شرایط دناچوراسیون آن از بالا به پایین در حال افزایش است (غلظت اوره و فرمامید از بالا به پایین ژل افزایش مییابد) قرار داده میشود. در این حالت، دو قطعه DNA که در یک جفت باز با هم اختلاف دارند در یک نقطه از ژل دناچوره نمیشوند. وقتی مولکولها به سطح بحرانی دناتوره برسند، دو رشته DNA شروع به جدا شدن میکنند. این نقطه بهعنوان نقطه ذوب عمل میکند. هرگاه قطعه DNA دناچوره شود، سرعت حرکت آن در ژل بهشدت کاهش مییابد، زیرا دو تک رشته حاصل به کمک مولکول سورالن به یکدیگر متصل شده و حجم آنها افزایش مییابد، در نتیجه عبور این تک رشتهها از خلال ماتریکس ژل کند میشود. مولکول سورالن پس از اتمام PCR به انتهای مولکول DNA متصل میشود. اگر هر دو آلل ژن مشابه هم باشند، محصول PCR یک نوع DNA دو رشتهای خواهد داشت، در نتیجه طی آزمایش DGGE یک باند بر روی ژل مشاهده میشود. اگر دو آلل حاوی جهش بوده و جهش در هر دو آلل یکسان باشد، محصول PCR شامل یک نوع DNA دو رشتهای موتانت میباشد. در این حالت نیز در ژل الکتروفورز یک باند دیده میشود، اما محل این باند با باند DNA سالم میتواند متفاوت باشد. اگر دو آلل یک ژن از لحاظ جهش نقطهای با هم تفاوت داشته باشند، محصول PCR بهصورت هترودوپلکس خواهد بود. این هترودوپلکسها پس از اتمام PCR و با حرارت دادن محصول واکنش بوجود میآیند.
شیب غلظت عامل دناچورهکننده باید با توجه به توالی قطعه مورد بررسی تعیین شود و در اصطلاح با توالی قطعه سازگار باشد. قطعات استانداردی وجود دارد که با استفاده از آنها میتوان شرایط محیطی را کنترل و تنظیم نمود.
در تکنیک DGGE نیازی به مواد شیمیایی سمی و یا رادیواکتیو نیست، ولی ابزارهای پیشرفته لازم است. نوع مشابهی از این روش به نام TGGE وجود دارد که در آن از حرارت بهجای غلظت مواد شیمیایی در ژل استفاده میشود. برای واسرشت شدن کامل قطعات حاصل از PCR، از یک ناحیه مصنوعی حاوی بالاترین دمای ذوب به نام clamp استفاده میشود.
آزمایش کوتاه شدن پروتئین
آزمایش کوتاه شدن پروتئین(PTT) برای یافتن جهشهایی طراحی شده است که طی آن اندازه پروتئین کدشده توسط ژن کوتاه میشود (جهشهایی مثل ایجاد کدون خاتمه، تغییر چارچوب و حذف قسمتی از ژن بهخصوص در ژنهای بزرگ).
این روش شامل یک RT-PCR با استفاده از دو پرایمر میباشد. پرایمر منفی موجب ساخته شدن رشته شده و پرایمر مثبت در انتهای ′۵ خود پروموتر فاژ T۷ را به همراه دارد. قطعه RNA به روش RT-PCR تکثیر شده و بهصـــــورت in vitro ترجمه میشود. قطعات ساختهشده، به یک سیستم رونویسی/ترجمه in vitro منتقل میشوند. این سیستم که معمولاً رتیکولوسیت است، حاوی tRNA و اسیدآمینه میباشد. بعضی از اسیدآمینههای این سیستم توسط رادیواکتیو نشاندار شدهاند. بعد از ساخت پپتید، محلول موجود در لوله بر روی ژل پلیاکریلامید حاوی SDS قرار داده میشود. الکتروفورز انجام شده و پپتید بر روی غشای نیتروسلولزی منتقل میشود. در این مرحله وسترن بلات و اتورادیوگرافی انجام شده و اندازه پپتید حاصل با پپتید استاندارد و سالم مقایسه میشود (شکل ۱). این تکنیک بهطور گسترده برای بررسی جهش در ژنهای (Adenomatous Polyposis Coli )APC، MSH۲، MLH۱ و دیستروفین بهکار میرود.
الف:
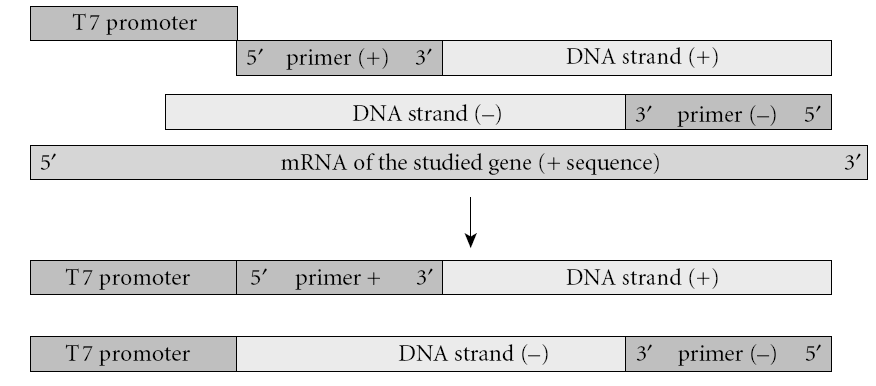
ب:
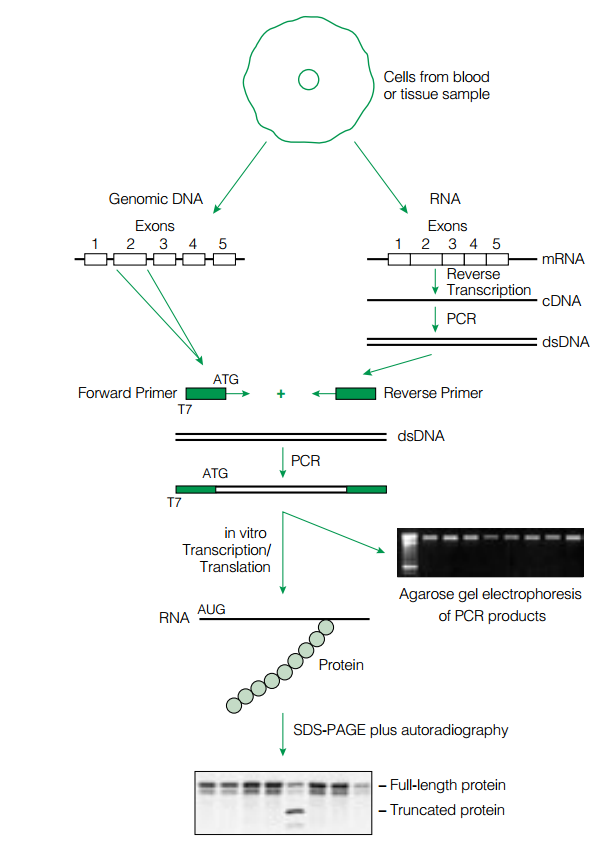
شکل ۱- اساس تکنیک PTT
الف:mRNA ژن موردنظر با کمک پرایمرهای (+) و (-) و استفاده از تکنیک RT-PCR تکثیر میشوند. قطعه بین دو پرایمر که در این مرحله حاوی پروموتر T۷ میباشد، وارد سیستم رونویسی/ ترجمه میشوند. در این سیستم، از tRNA حاوی آمینواسید نشاندار استفاده میشود.
ب: پیپیدهای ساختهشده، با استفاده از SDS-PAGE، جدا شده و به روی غشاء منتقل میشوند. با استفاده از وسترن بلات و اتورادیوگرافی، اندازه قطعات تعیین و با حالت استاندارد مقایسه میشود
برش شیمیایی فاز جامد
برش شیمیایی نوکلئوتیدهای متصلنشده (CCM) یکی از روشهای انتخابی در تشخیص جهش میباشد. این روش قادر به شناسایی تمام جفت نوکلئوتیدهای متصلنشده و تکرشتهای میباشد. نوکلئوتیدهای متصلنشده، ناپایدار بوده و بهشدت مستعد واکنش آنزیمی و شیمیایی میباشند. در این تکنیک ابتدا از دو ماده شیمیایی هیدروکسیلآمین و پرمنگنات پتاسیم، بهمنظور واکنش با بازهای سیتوزین و تیمین تک رشتهای استفاده میشود، سپس نمونه با پیپریدین مجاور شده و بازهای ناجور که در مرحله قبل تغییر یافتهاند، بریده میشوند. قطعات حاصل از برش مرحلۀ قبل، بر روی ژل پلیاکریلامید دناچوران الکتروفورز میشود تا مکان بازهای جهشیافته مشخص شود (شکل ۲). در طول زمان، این روش تغییراتی کرده است و در نتیجه مزایایی نیز کسب کرده است که میتوان به موارد زیر اشاره نمود:
- پرمنگنات پتاسیم (KMnO۴) جایگزین ماده سمی تترا اکسید اسمیوم (OsO۴) شده است.
- اگر هر دو نمونه DNA وحشی و جهشیافته نشاندار شوند، شانس شناسایی جهش دو برابر خواهد شد.
- این روش حساس بوده و با مقدار نمونه کمتر از ۰/۱ میکروگرم نیز میتوان جهش را شناسایی کرد.
- همه مراحل آزمایش بر روی سطح جامد ذرات سیلیکونی انجام میگیرد. در نتیجه برای تغییرات و دستکاریهای بعدی مناسب است.
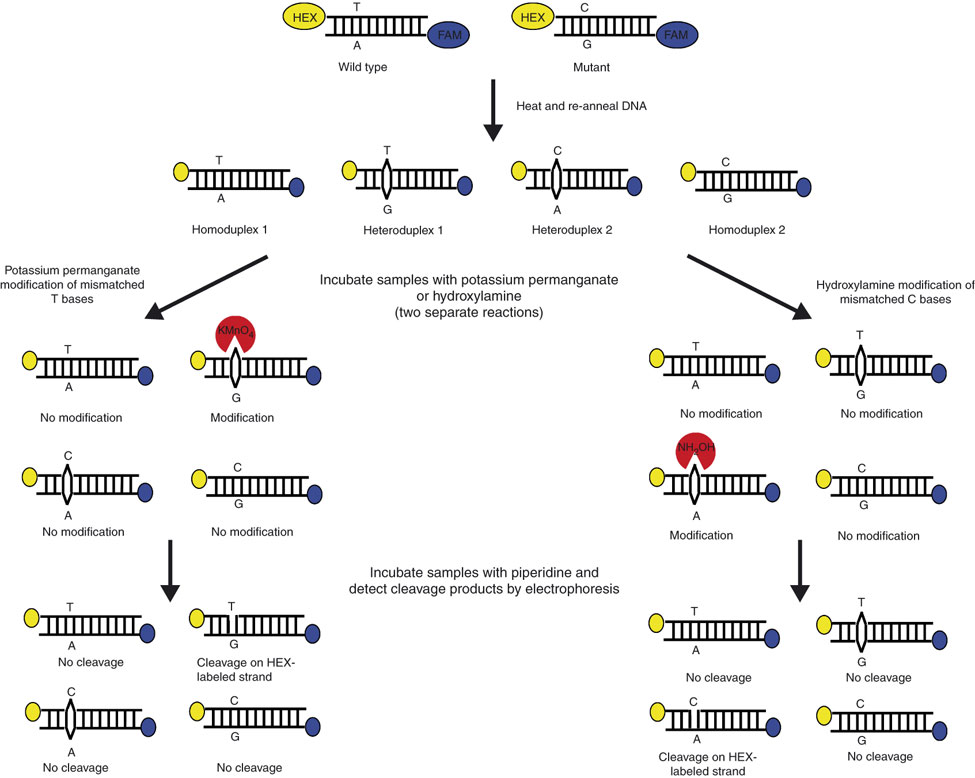
شکل ۲: تصویر شماتیک برش شیمیایی فاز جامد
دو نمونه طبیعی و جهشیافته به کمک آغازگر مستقیم نشاندار شده با HEX (زرد) و آغازگر معکوس نشاندار شده با ۶-FAM (آبی) تکثیر شده و از آنها هترودوپلکس تهیه میشود. هترودوپلکس حاصل بر روی فاز جامد سلیکا تثبیت شده و به کمک واکنشهای شیمیایی تغییر میکند. یکی از این نمونهها با هیدروکسیل آمین (بهمنظور تغییر باز ناجور C) و دیگری با پرمنگنات پتاسیم (بهمنظور تغییر شیمیایی باز ناجور T) تیمار میشوند. پیپریدین بازهای تغییریافته را برش داده و رشته DNA از فاز جامد جدا شده و بهصورت کاپیلاری الکتروفورز میشود
این تکنیک بر اساس تشکیل هترودوپلکس بنا نهاده شده است، بنابراین بهمنظور انجام آزمایش، دو قطعه مکمل نمونه و کنترل را با هم مخلوط کرده، ذوب میکنند. دوباره با کاهش دما، این دو رشته به هم متصل میشوند. اگر توالی این دو قطعه با هم یکسان نباشد، بر روی هترودوپلکس حاصل، باز متصل نشده و تکرشتهای قرار میگیرد. بازهای ناجور C,T مستعد تغییرات و برش شیمیایی میباشند. از آنجا که همۀ انواع جفتهای ناجور C,T(CC, CT, CA, TT, TG, TC) در این روش شناسایی و بریده میشوند، پس با استفاده از DNA کاوشگر کنترل (وحشی) و جهشیافته، میتوان همۀ جهشهای نقطهای را غربالگری نمود. بهمنظور دو برابر شدن احتمال شناسایی جهش، بهتر است که هر دو نمونه وحشی و جهشیافته را نشاندارکرد. کاوشگر DNA را میتوان بهصورت اولیگونوکلئوتید مصنوعی ساخت و یا با استفاده از PCR از ژنوم موجود زنده به دست آورد.
مواد لازم برش شیمیایی فاز جامد:
۱) TE buffer:
۱۰۰µl of ۱ M Tris-HCl (pH ۸,۰)
۲۰ µl of ۰,۵ Methylenediamine-tetraacetic acid (EDTA)
۹٫۸۸ mL of distilled water
Store the TE buffer at room temperature
۲) ۴٫۲ M Hydroxylamine solution:
۱٫۳۹ g solid hydroxylamine hydrochloride is dissolved in ۱,۶ mL distilled water
این محلول را با حدود ۱ سیسی دی اتیل آمین به pH=۶ برسانید.
Distilled water to ۴ml
Store at –۲۰°C for up to ۶ months
۳) ۳ M tetraethylammonium chloride (TEAC) solution:
۴۹٫۷ g of tetraethylammonium chloride is dissolved in ۱۰۰ mL distilled water. Store at ۴°C for up to ۳ months.
۴)۱ mM KMnO۴ solution:
۸۰mg of KMnO۴ is dissolved in ۵ mL of distilled water.
µl۱۰ از این محلول را با µl۹۰۰ از محلول ۳ مولار TEAC مخلوط کنید تا محلول ۱ میلیمولی از KMnO۴ بدست آید. این محلول باید تازه باشد.
۵) Cleavage-dye solution:
۲۰µl undiluted Piperidine
۶۴µl formamide
۱۶µl dye (۵۰ mg blue dextran per mL)
Store at ۴°C for ۱ d.
۶) Fluorophore ۶-FAM and HEX for the ۵′ and ۳′ primers
۷) Tris-borate EDTA (TBE) buffer for electrophoresis (pH = ۸,۰):
۱۶٫۲ g Tris base,
۸٫۱ g boric acid,
۱٫۱۲ g EDTA
۱۵۰۰ mL distilled water,
Store at ۲۵°C.
۸) کیت PCR
۹) کیت تخلیص محصول PCR
۱۰) بستر جامد (Solid support for DNA)
روش انجام کار:
همه مراحل کار باید در زیر هود انجام شود، زیرا مواد مورد استفاده در این آزمایش از قبیل اکریلامید، هیدروکسیل آمین، فرمامید و پیپریدین سمی هستند.
آمادهسازی نمونه:
قطعه موردنظر را با استفاده از پرایمرهای نشاندار شده با فلوروفور PCR نمایید، سپس قطعه حاصل را با کمک کیت تخلیص محصول PCR یا به روش برش باند موردنظر از ژل، جدا کنید. غلظت DNA را با کمک جذب UV در nm۲۶۰ یا بر روی ژل محاسبه کنید.
تشکیل هترودوپلکس:
مقدار مساوی از DNA وحشی و جهشیافته را برداشته و در بافر TE مخلوط نمایید. محلول را در ترموسایکلر قرار داده، به مدت ۷ دقیقه ۹۹ درجه سلسیوس گرما داده و سپس دما را به ْ۶۵ رسانده و یک ساعت در این دما نگهدارید. در نهایت به مدت ۳۰ دقیقه در ۲۵ درجه سلسیوس قرار دهید تا محلول خنک شود.
اتصال هترودوپلکس و همودوپلکس به ذرات سیلیکونی:
- µl۱ از DNA هترودوپلکس را به درون دو میکروتیوب بریزید (یک میکروتیوب برای واکنش با پرمنگنات پتاسیم و دیگری برای واکنش هیدروکسیل آمین).
- µl۱ از DNA همودوپلکس را به درون دو میکروتیوب بریزید (یک میکروتیوب برای واکنش با پرمنگنات پتاسیم و دیگری برای واکنش هیدروکسیل آمین).
- µl۲/۵ از سوسپانسیون اتصال ذرات سیلیکونی را به هرکدام از میکروتیوبها اضافه کرده و ۲-۱ ساعت در دمای اتاق، بر روی شیکر قرار دهید.
- با استفاده از µl۲۰۰ محلول شستشو، ذرات حاوی DNA را دو بار شستشو دهید.
- ذرات را به مدت ۱۵ دقیقه در هوای آزاد و در دمای ۲۵ درجه سلسیوس قرار داده تا خشک شوند.
واکنش با پرمنگنات پتاسیم:
µl۳۰ از محلول mM۱ پرمنگنات در ۳ مولار TEAC را به درون دو میکروتیوب مربوط به پرمنگنات پتاسیم ریخته و به مدت ۱۰ دقیقه در ۲۵ درجه سلسیوس انکوبه کنید. میکروتیوبها را در g۳۲۵ سانتریفیوژ کرده و محلول رویی را بهآرامی با پیپت پاستور بردارید. سپس رسوب را دو بار با µl۲۰۰ محلول شستشو، شسته و در ۱۵ دقیقه در هوای آزاد خشک نمایید.
واکنش با هیدروکسیل آمین:
µl۳۰ از محلول ۴/۲ مولار هیدروکسیل آمین در TEAC را به درون دو میکروتیوب مربوطه ریخته و به مدت ۴۰ دقیقه در ۳۷ درجه سلسیوس انکوبه کنید. میکروتیوبها را سانتریفیوژ کرده و محلول رویی را دور بریزید. سپس رسوب را دو بار با µl۲۰۰ محلول شستشو، شسته و در ۱۵ دقیقه در هوای آزاد خشک نمایید.
برش با پیپریدین:
µl۱۰ از رنگ برش (Cleavage-dye) را به هر یک از چهار میکروتیوب اضافه کرده و به مدت ۳۰ دقیقه در دمای ۹۰ درجه سلسیوس قرار دهید. میکروتیوبها را بر روی یخ، خنک نموده و سانتریفیوژ کنید. محلول رویی را بر روی ژل پلیاکریلامید دناچوران ۴/۲۵% حاوی ۶ مولار اوره، با بافر TBE الکتروفورز نمایید. برای یک قطعه به طول ۵۰۰ جفت باز، شرایط الکتروفورز شامل ۳۰۰۰ ولت، به مدت ۳ ساعت میباشد.
شناسایی جهش در این روش بر اساس مقایسه باندهای ژل همودوپلکس با ژل هترودوپلکس میباشد. اگر باندی بر روی ژل هترودوپلکس وجود داشته باشد، ولی در ژل همودوپلکس دیده نشود، نشاندهنده وجود جهش در نمونه میباشد (شکل ۳).
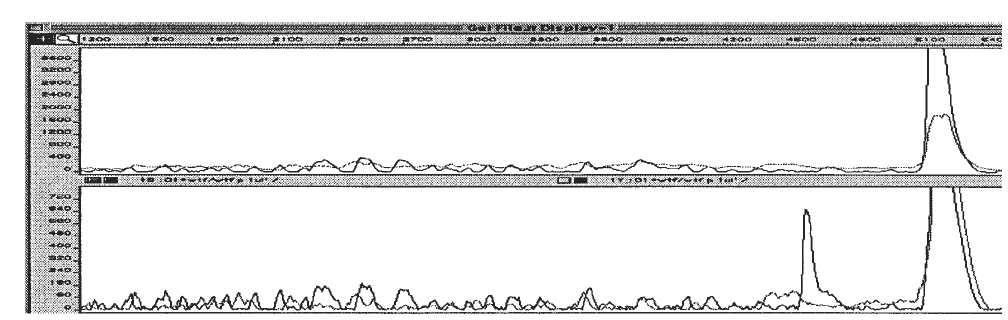
شکل ۳: شناسایی جفت باز ناجور TC در یک قطعه ۵۴۰ نوکلئوتیدی به روش برش شیمیایی
در بالای شکل منحنی مربوط به نمونه کنترل دیده میشود که قله مربوط به برش شیمیایی وجود ندارد، اما در منحنی پایین، قله مربوط به باز ناجور T بهطور قوی دیده میشود
نکات قابلتوجه:
- بر اساس واکنشپذیری بازهای ناجور RNA و DNA، سه تکنیک به وجود آمده است: روش برش غیر رادیواکتیو با استفاده از ریبونوکلئاز A، برش آنزیمی باز ناجور (EMC) و نهایتاً برش شیمیایی باز ناجور (CCM).
روش آنزیمی برش، بهصورت کیت در دسترس است و در آن آنزیم اندونوکلئاز VII فاژ T۴ جفت بازهای ناجور را برش میدهد. از مزایای روش آنزیمی این است که واکنش در یک مرحله انجام میشود. درحالیکه در روش شیمیایی واکنش دو مرحلهای است. همچنین در روش آنزیمی، یک آنزیم همه انواع بازهای ناجور را شناسایی میکند، اما در روش شیمیایی دو ماده شیمیایی موردنیاز است.
از معایب روش آنزیمی میتوان به مواردی از قبیل گران بودن روش، نیاز به بهینهسازی تمام شرایط واکنش و وجود باندهای زمینهای فراوان که ناشی از برش بازهای جور میباشد، اشاره کرد.
در روش برش با ریبونوکلئاز، به تولید RNA نیاز است. از آنجا که قطعات بریدهشده در این روش، دو رشتهای هستند، نتیجه واکنش را بر روی ژل آگاروز میتوان مشاهده کرد.
- در تکنیکهای برشی تاکنون مثبت و منفی کاذب گزارش نشده است، با این وجود بهتر است که هر دو نمونه حاوی جهش و کنترل، نشاندار شوند تا اگر جهشهای نادر غیرواکنشینیز وجود دارند، شناسایی شود. همچنین در این تکنیکها میتوان از مواد رادیواکتیو بهجای فلوروفور، برای نشاندار کردن استفاده نمود.
- در این تکنیک، اندازه، غلظت و دمای ذوب هترودوپلکس باید مدنظر قرار گیرد؛ بهعنوان مثال برای تشکیل هترودوپلکس در قطعۀ غنی از C-G، دمای ۱۰۰ درجه سلسیوس باید داده شود. بعد از تشکیل هترودوپلکس باید نمونه بر روی آگاروز الکتروفورز شده و بررسی شود که چندین باند قوی در ژل وجود نداشته باشد؛ یا نمونه در ژل اسمیر نشده باشد.
- اتصال دوپلکسهای DNA به ذرات سیلیکونی جزء مهمترین قسمت آزمایش میباشد. این اتصال در شرایط نمکی حاوی ۳ مول TEAC رخ داده و در نتیجه در هنگام تغییرات شیمیایی DNA و مراحل شستشو این اتصال باقی خواهد ماند.
- اندازۀ قطعه مورد آزمایش توسط عواملی از قبیل مشکلات تکنیکی، صحت تشکیل هترودوپلکس و بستر جامد (ذرات سیلیکونی) محدود میشود. با استفاده از بستر سیلیکونی قطعات تا ۵۰۰ بازی را میتوان آزمایش کرد. برای قطعات بزرگتر از فاز مایع باید استفاده نمود.
- در اصل علاوه بر TEAC از مواد دیگری نیز میتوان استفاده کرد، اما TEAC نسبت به دیگر مواد، از سمیت کمتری برخوردار است، همچنین این ماده بهتر با DNA واکنش نشان میدهد. در این روش TEAC بهعنوان ناپایدارکننده دوپلکس عمل کرده و واکنش نوکلئوتیدها را با هیدروکسیلآمین و پرمنگنات پتاسیم افزایش میدهد.
- محلول پرمنگنات پتاسیم باید تازه باشد. اگر این محلول یک روز بماند به رنگ زرد- قهوهای درآمده و MnO۲ رسوب خواهد کرد. واکنش به دما و غلظت سوبسترا بستگی دارد.
- انکوباسیون طولانیمدت میتواند منجر به واکنش اضافی شده و هترودوپلکس و قطعات DNA را تخریب نماید. انکوباسیون کوتاهمدت نیز مانع واکنش خواهد شد.